
 On February 13, 2017, the FDA’s reclassification of rapid antigen influenza testing became effective. The reclassification impacts manufacturers of certain diagnostic devices used to test for influenza (flu).
On February 13, 2017, the FDA’s reclassification of rapid antigen influenza testing became effective. The reclassification impacts manufacturers of certain diagnostic devices used to test for influenza (flu).
Currently, there are a number of diagnostic tests that can help in the diagnosis of flu. They break down into two broad categories: Point-of-Care-based and Laboratory-based.
In the Point-of-Care Testing (POCT) space, two common techniques are:
- Rapid Immunochromatographic Detection Tests (RIDTs), including both digital reader systems & traditional, manual read assays.
- Rapid Molecular Tests
In larger laboratories and reference labs, you may find:
- Immunofluorescence Direct or Indirect Antibody Staining
- RT-PCR (nucleic acid methods are the new gold standard, replacing the cell culture)
- Rapid Cell Culture
- Viral Tissue Cell Culture
RIDT Tests in the Point-of-Care Setting
Rapid Influenza Diagnostic Tests (RIDTs) are immunoassays that can identify the presence of influenza A and B viral nucleoprotein antigens in respiratory specimens, with results displayed qualitatively. There are a number of commercially-available RIDTs in use in the U.S.

RIDT tests are frequently used in point-of-care settings due to their cost-effectiveness, ease-of-use, and rapid results (often available in 15 minutes or less). These tests give doctors the ability to begin antiviral therapy (while reducing antibiotic use) during a single patient visit – ultimately leading to a decrease in hospitalizations and fewer unnecessary tests.
FDA Tightens Standards, Reclassifies RIDTs
However, there have historically been some challenges with RIDTs and their variability in performance. While they tend to show good specificity, they can deliver less-than-optimal sensitivity in respiratory specimens compared to RT-PCR or viral culture.
In some cases (for example, the H1N1 flu strain in 2009) RIDTs may be unable to detect the virus at all. For this reason, negative RIDT test results should be interpreted with caution given the potential for false negative results - especially during peak flu season in a community.
Based on these concerns, the FDA began work to reclassify antigen-based RIDTs as Class II devices in order to improve the overall quality of testing for influenza. The FDA has provided certain performance criteria that manufacturers will have to meet in order to market their influenza assays.
The new standards went into effect in February 2017. According to the FDA:
“The new minimum performance requirements for these tests detecting influenza virus antigens are expected to lower the number of misdiagnosed influenza infections by increasing the number of devices that can reliably detect the influenza virus.”
What Does the FDA Reclassification Mean for RIDT Manufacturers?
For manufacturers who currently have a marketed device that does not meet the minimum performance criteria, a new 510(k) must be submitted demonstrating compliance with the special controls included in the new clinical performance standards before marketing the device.
There are several key takeaways from the FDA’s move:
- Rapid Antigen Influenza tests have been reclassified from Class I to Class II medical devices.
- Minimum performance requirements are mandated as compared to viral culture or molecular methods.
| VIRAL CULTURE | MOLECULAR | |||
| VIRUS | Sensitivity | Specificity | Sensitivity | Specificity |
| INFLUENZA A | 90% (80% CI*) | 95% (90% CI) | 80% (70% CI) | 95% (90% CI) |
| INFLUENZA B | 80% (70% CI) | 95% (90% CI) | 80% (70% CI) | 95% (90% CI) |
*CI-Lower Confidence Interval
- Annual and Emergency Analytical special controls testing (test panel) will be undertaken by manufacturers to ensure continued performance monitoring. The FDA will collaborate with the Centers for Disease Control and Prevention (CDC) to ensure that an influenza virus analytical reactivity test panel is available to all test manufacturers.
- By January 12, 2018, manufacturers are required to be in compliance with the requirements imposed by the FDA to meet the Class II standards for rapid influenza diagnostic testing.
What Does the FDA Reclass Mean for Physicians and Labs?
Facilities testing for flu can continue to purchase their current product up until January 12, 2018 and utilize them until the kit’s expiration date. This provides facilities enough time to assess the performance data against the FDA criteria and engage clinicians and management on the next course of action for flu testing.
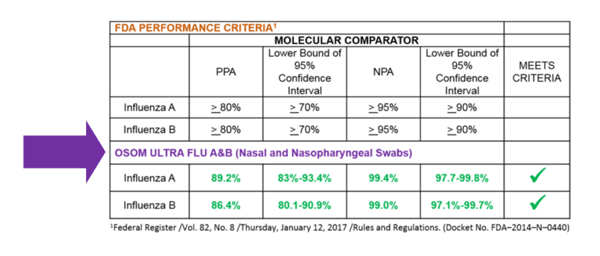
OSOM® Ultra Flu A&B Meets New FDA Criteria
We know you’re concerned about diagnosing patients correctly – especially during flu season, when so many symptoms can mimic other diseases.
Sekisui Diagnostics is committed to ensure that our products comply with the requirements imposed by the FDA to meet the Class II standards for rapid influenza diagnostic testing.
The OSOM® Ultra Flu A&B (Catalog #1006, view package insert) meets the performance criteria defined in the final rule (21 CFR 866.3328).
Questions about a Sekisui Diagnostics’ Rapid Influenza Diagnostic Test? Give us a call!



Share Article